Leveraging AI for Regulatory Compliance in Life Sciences

The Evolution of Digital Integrity in Clinical Research
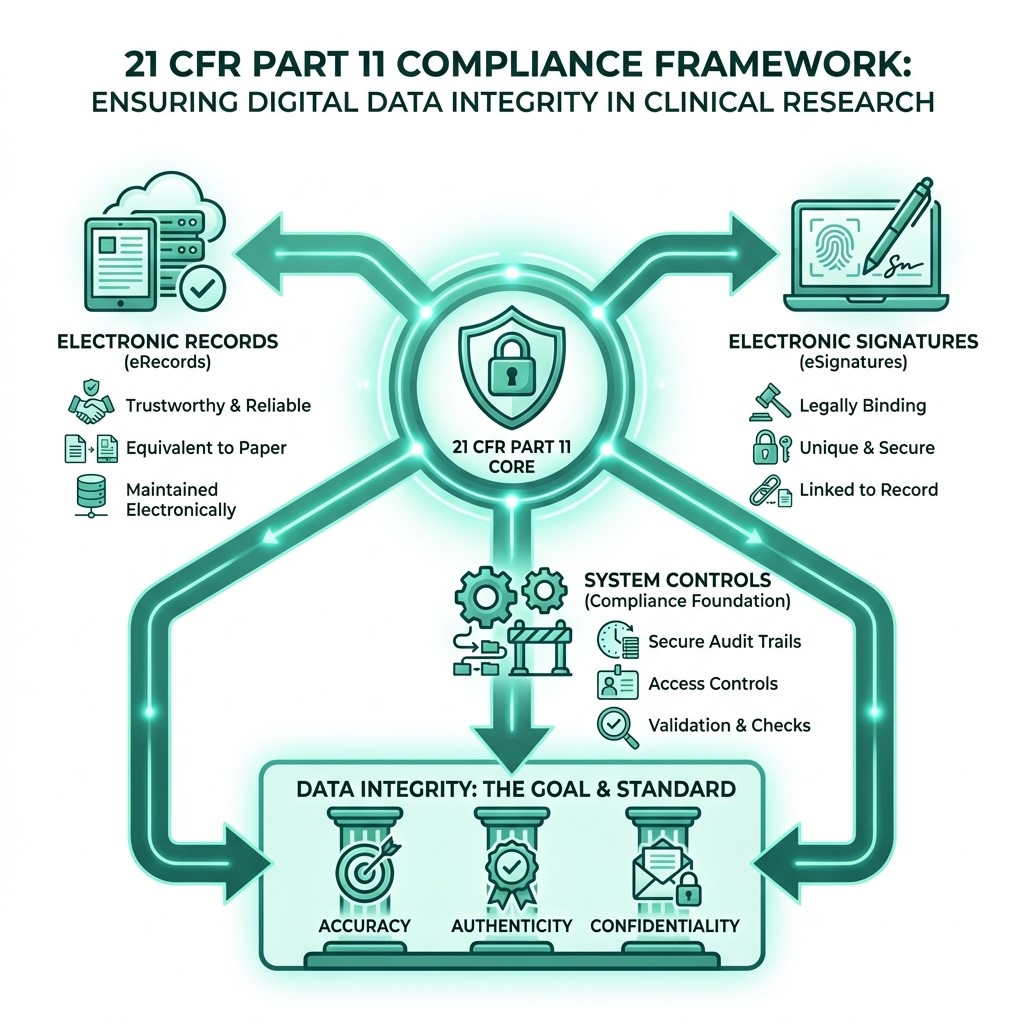
As clinical research embraces digital transformation, the importance of regulatory compliance has never been greater. The FDA’s 21 CFR Part 11 stands as a cornerstone, setting the gold standard for electronic records and signatures. It ensures their trustworthiness and reliability in an increasingly paperless environment.
Navigating its intricate requirements can be a significant challenge for sponsors, Contract Research Organizations (CROs), and research sites alike. We understand the complexities involved in maintaining data integrity while leveraging cutting-edge technology.
In this comprehensive guide, we will delve into the core tenets of 21 CFR Part 11. We will explore its purpose, scope, and the critical distinction between ‘Part 11 ready’ and ‘fully compliant’ systems. We will also examine the nuances of electronic record management, signature validation, and the evolving landscape of computer software assurance.
Furthermore, we will shed light on how innovative approaches, including AI-powered life sciences compliance, are revolutionizing how organizations achieve and maintain regulatory compliance. Join us as we demystify Part 11 and equip you with the knowledge to thrive in the digital age of clinical research.
A continuous quest for efficiency, accuracy, and patient safety has marked the journey of clinical research. With the advent of digital technologies, paper-based processes are rapidly giving way to electronic systems, promising faster data collection, improved accessibility, and reduced administrative burden. However, this digital transformation introduces new challenges, particularly in ensuring the integrity and reliability of electronic data. This is where 21 CFR Part 11 becomes indispensable.
Enacted by the FDA in 1997, 21 CFR Part 11 was a landmark regulation designed to address the growing use of electronic records and electronic signatures (ERES) in FDA-regulated industries. Its primary purpose is to establish the criteria under which the FDA considers ERES to be trustworthy, reliable, and equivalent to paper records and handwritten signatures. In clinical research, this means that data captured electronically, from patient demographics to adverse event reporting, must meet stringent standards to be accepted by regulatory authorities. The regulation ensures that as we move towards a fully digital ecosystem, the fundamental principles of data integrity – accuracy, authenticity, and confidentiality – are upheld.
Understanding 21 CFR Part 11 in Modern Life Sciences Compliance
At its core, 21 CFR Part 11 is about ensuring that electronic records and electronic signatures are as legally sound and reliable as their paper counterparts. It applies to any records required by the FDA under predicate rules that are maintained electronically rather than on paper. This includes a vast array of documents and data generated throughout the clinical research lifecycle, including study protocols and informed consent forms, case report forms (CRFs), laboratory data, and regulatory submissions.
The regulation outlines general provisions, requirements for electronic records, and specific controls for electronic signatures. It mandates that systems that generate, process, or store these electronic records must have controls to ensure their authenticity, integrity, and confidentiality. This includes features like secure, time-stamped audit trails, operational system checks, and robust security measures to prevent unauthorized access or alteration. For a deeper dive into the official text, you can consult the eCFR for 21 CFR Part 11.
One critical distinction in Part 11 concerns the type of electronic system used: closed versus open.
Closed Systems: These are systems in which access to electronic records is controlled by the persons responsible for their content. In clinical research, this often refers to proprietary systems managed by a sponsor or CRO, where the sponsor or CRO has direct control over user access, system configuration, and data management. The controls for closed systems, outlined in § 11.10, focus on ensuring the integrity and authenticity of records through measures like validation, audit trails, and access controls.
Open Systems: These are systems in which access to electronic records is not controlled by the persons responsible for their content. Examples might include cloud-based services or systems accessible over public networks. Because the environment is less controlled, Part 11 requires additional measures for open systems to ensure record integrity and authenticity from creation to receipt. This often involves encryption, digital certificates, and other advanced security protocols.
Understanding this distinction is vital for selecting and implementing compliant technology in clinical trials.
Feature Closed Systems Open Systems ControlAccess controlled by content creators/responsible parties Access not controlled by content creators/accountable parties (e.g., public networks) Risk Profile Generally lower, as control is centralized Generally higher, due to external accessibility Key Requirement Validation, audit trails, access controls, system checks Additional measures like encryption, digital signatures, integrity checks Examples Internal EDC systems, sponsor-managed eTMF Cloud-based platforms, systems accessed via public internet The Role of Predicate Rules in Life Sciences Compliance
21 CFR Part 11 does not exist in a vacuum. Its applicability is directly linked to what are known as “predicate rules.” These are the underlying FDA regulations that requirespecific records to be maintained. If a record is mandated by a predicate rule (e.g., 21 CFR Part 312 for Investigational New Drug Applications or 21 CFR Part 58 for Good Laboratory Practice), and an organization chooses to keep that record electronically, then 21 CFR Part 11 applies. If a predicate rule does not require a record, or if an organization decides to keep a paper copy as the official record, then Part 11 generally does not apply to the electronic version of that record.
This distinction is crucial because it helps organizations narrow the scope of their Part 11 compliance efforts. For example, while 21 CFR Part 11 sets the framework for electronic records and signatures, other regulations, such as the Federal Food, Drug, and Cosmetic (FD&C) Act and the Public Health Service (PHS) Act, serve as the “predicate rules” that dictate which records must be kept. The FDA’s guidance clarifies that Part 11 applies to documents that are created, modified, maintained, archived, retrieved, or transmitted under any records requirements outlined in agency regulations. For more on this, the FDA provides extensive information on Part 11 compliance FAQs, emphasizing the role of predicate rules.
Distinguishing Between Part 11 Ready and Fully Compliant Systems
A common source of confusion in the industry is the difference between a system being “Part 11 ready” and “fully Part 11 compliant.” Many software vendors market their products as “Part 11 ready,” implying that the technology itself has the necessary features to support compliance. This usually means the system includes technical specifications such as audit trails, security features, and electronic signature capabilities.
However, “Part 11 ready” is not the same as “fully compliant.” Full compliance extends beyond the software’s technical features and encompasses the entire system, including the organization’s processes, personnel, and documentation. As the FDA guidance emphasizes, compliance requires a holistic approach. A “Part 11 ready” system only provides the tools; it is up to the user organization to implement the procedural controls, develop Standard Operating Procedures (SOPs), conduct personnel training, and perform the necessary system validation to achieve full compliance.
Therefore, when evaluating technology for clinical research, recognize that no system comes “pre-validated” or “pre-compliant” out of the box. Validation is an activity performed by the user organization, often in collaboration with the vendor. While vendors play a crucial role by providing comprehensive documentation, technical specifications, and, in some cases, validation support packages, the ultimate responsibility for compliance lies with the regulated entity (sponsor, CRO, or site). Organizations must generate documentation defining platform requirements and conduct documented testing to ensure the system performs as intended within their specific operational context. This is a key point highlighted in the FDA’s guidance on the scope and application of Part 11.
Transitioning from Traditional Validation to Computer Software Assurance (CSA)
Historically, software validation in regulated environments, including clinical research, has been a rigorous, documentation-heavy process often following a V-model approach. This traditional approach, while thorough, could be time-consuming and resource-intensive, sometimes outweighing the actual risk posed by the software. Recognizing these challenges and aiming to encourage the adoption of innovative technologies, the FDA has been actively promoting a shift towards Computer Software Assurance (CSA).
CSA represents a more risk-based approach to software validation, moving away from extensive documentation for low-risk systems and focusing instead on critical thinking and assurance activities. The goal is to establish confidence in software quality and fitness for its intended use, with documentation proportionate to the risk. This aligns with the FDA’s “General Principles of Software Validation,” which encourages a risk-based approach. The FDA issued its final Computer Software Assurance for Production and Quality System Software guidance in September 2025 (replacing the 2022 draft), establishing this risk-based framework. This new guidance aims to streamline the validation process, making it more efficient without compromising patient safety or data integrity.
This evolution is particularly relevant for clinical research, where a wide range of software platforms is used, from Electronic Data Capture (EDC) systems to Clinical Trial Management Systems (CTMS) and eTMF solutions. Not all technology platforms used in clinical research require the same level of rigorous validation. A risk-based approach dictates that the validation effort should be commensurate with the potential impact of the software on patient safety, data integrity, and product quality. For example, a system directly involved in collecting primary efficacy endpoints would warrant more extensive assurance activities than a system used for scheduling non-critical meetings.
This is where advanced solutions, such as AI-powered life sciences compliance solutions, come into play. AI can automate parts of the assurance process, identify potential risks, and generate risk-based testing scenarios, reducing the manual burden and accelerating time to compliance. This paradigm shift is reflected in industry trends: a recent study found that 58% of companies are already using digital validation systems, with another 35% planning to adopt them over the next 2 years.
Scaling AI-Powered Life Sciences Compliance Across Global Sites
Clinical trials are increasingly global, involving multiple sites, diverse regulatory landscapes, and a complex web of sponsors, CROs, and vendors. Scaling compliance efforts across such a distributed environment presents significant challenges. Ensuring the consistent application of 21 CFR Part 11, alongside other international regulations such as ICH GCP, across all participating entities requires robust systems and processes.
AI-powered compliance solutions can significantly aid in this scaling. They can help harmonize data collection and management practices across sites, automate the review of electronic records for compliance, and provide real-time insights into the compliance status of systems and processes. This reduces variability, enhances data quality, and ensures that all electronic records and signatures, regardless of origin, meet regulatory standards. Such tools can also facilitate automated auditing and provide regulatory intelligence, adapting to local requirements while maintaining overarching global compliance.
Managing Legacy Systems and Enforcement Discretion
The FDA recognizes that many organizations operate “legacy systems” – older electronic systems that were in use before the August 20, 1997, effective date of 21 CFR Part 11. While the original regulation applied broadly, the FDA issued guidance in 2003 clarifying its enforcement discretion. This guidance indicated that, for legacy systems, the FDA would exercise discretion regarding particular Part 11 requirements (such as specific validation elements or audit trail features), provided that the system was deemed “fit for its intended use” and that predicate rule requirements were still met.
This enforcement discretion was a response to industry concerns about the cost and impracticality of retrofitting older systems to meet every granular detail of Part 11. However, it’s crucial to understand that this discretion does not absolve organizations of their responsibility to ensure data integrity and compliance with predicate rules. Organizations must document their assessment of legacy systems, demonstrate their fitness for use, and implement appropriate controls to mitigate risks. Any system upgrades or replacements, however, would typically be expected to meet the whole Part 11 requirements. The FDA’s guidance on Part 11 Scope and Application provides further details on this aspect of enforcement.
Implementing AI-Driven Controls for Electronic Records and Signatures
The core of 21 CFR Part 11 lies in the specific controls required for electronic records and electronic signatures. These controls are designed to ensure the trustworthiness, reliability, and equivalence of ERES to paper records and handwritten signatures. We can categorize these into requirements for electronic records and specific controls for electronic signatures.
For electronic records, key requirements include:
- Validation: Systems must be validated to ensure accuracy, reliability, consistent intended performance, and the ability to discern invalid or altered records. This is a fundamental aspect of Part 11 compliance.
- Audit Trails: Secure, computer-generated, time-stamped audit trails must be maintained to record all actions related to electronic records, including creation, modification, and deletion. These trails must be available for agency review and should not obscure previous entries.
- Access Controls: Limiting system access to authorized individuals is paramount. This involves unique user IDs, passwords, and other authentication methods to prevent unauthorized access, changes, or deletions.
- Record Retention: Electronic records must be retained for the period specified by predicate rules and be readily retrievable. This also includes ensuring the ability to generate accurate, complete copies in both human-readable and electronic formats.
- System Checks: Operational system checks to enforce the sequence of steps, date and time stamping, and other controls are necessary.
- Copying and Archiving: The ability to generate accurate, complete copies of records in both human-readable and electronic formats, suitable for inspection, review, and copying, is required.
AI can significantly enhance these controls. For instance, AI-driven anomaly detection can monitor audit trails for suspicious activities, flagging potential data integrity breaches in real-time. Machine learning algorithms can also assist in risk-based validation by identifying critical functionalities that require more rigorous testing.
Technical Requirements for Compliant Electronic Signatures
Electronic signatures are perhaps the most distinctive aspect of 21 CFR Part 11. To be considered legally equivalent to a handwritten signature, an electronic signature must meet several stringent requirements as outlined in § 11.50, § 11.70, § 11.200, and § 11.300 of the regulation. These include:
- Signature Manifestation: The electronic signature must include the printed name of the signer, the date and time of the signature, and the meaning (e.g., review, approval, authorship, responsibility). This ensures clarity and accountability.
- Signature/Record Linking: Electronic signatures must be securely linked to their respective electronic records to prevent them from being excised, copied, or otherwise transferred to falsify a record.
- Unique Identification: Each electronic signature must be assigned to a single individual and not reused or reassigned.
- Authentication: The identity of the individual signing electronically must be verified. This often involves a combination of two distinct components, such as an ID code and a password. Advanced methods like biometrics or two-step authentication (e.g., a password combined with a one-time password (OTP) sent to a registered device) are increasingly common and highly recommended for enhanced security. Practical examples, such as Adobe Sign, when configured with Bio-Pharma settings and two-step authentication, can meet these requirements for study-level documents in clinical research.
- Non-Repudiation: The system must ensure that the signer cannot later deny having signed the electronic record. This is why a paper non-repudiation letter to the FDA is sometimes required before an organization can begin using electronic records and signatures, even though it may seem counterintuitive in a digital age. As noted by one source, the primary purpose of Part 11 is to ensure electronic records are equivalent to paper records with handwritten signatures.
Operational Controls and System Validation Procedures
Beyond the technical features, operational controls and robust validation procedures are essential for maintaining 21 CFR Part 11 compliance. These include:
- System Validation: This is a continuous process that begins with the system’s development and continues throughout its lifecycle. It typically involves:
- Installation Qualification (IQ):Verifying that the system is installed correctly and according to specifications.
- Operational Qualification (OQ):Confirming that the system functions as intended across its operating range.
- Performance Qualification (PQ):Demonstrating that the system consistently performs its intended function under actual use conditions.
- The FDA’s “General Principles of Software Validation” provides detailed guidance on this.
- Standard Operating Procedures (SOPs): Comprehensive SOPs must be in place for all aspects of system operation, maintenance, security, and data management. This includes procedures for creating, managing, and using electronic signatures.
- Training: All personnel using the electronic system must receive adequate training on its operation, relevant SOPs, and the implications of 21 CFR Part 11.
- Change Control: A formal change control system is necessary to manage any modifications to the validated system. This ensures that changes are documented, tested, and do not compromise the system’s validated state or compliance.
- Certified Copies: Organizations must be able to generate certified copies of electronic records that wholly and accurately represent the original electronic data.
These operational controls, combined with the technical requirements, form a comprehensive framework for ensuring the integrity and reliability of electronic records and signatures in clinical research.
Best Practices for Maintaining a Compliant Digital Ecosystem
Achieving 21 CFR Part 11 compliance is not a one-time event; it’s an ongoing commitment to maintaining a robust and trustworthy digital ecosystem. Organizations must implement best practices that integrate compliance into their daily operations and technology lifecycle.
- Continuous Monitoring and Review:Regular monitoring of system performance, security logs, and audit trails is crucial. This proactive approach helps identify potential issues before they escalate into non-compliance.
- Vendor Management: When using third-party software or cloud services, thorough vendor qualification and ongoing oversight are essential. Organizations must ensure that their vendors understand and adhere to Part 11 requirements and provide necessary documentation to support the user’s compliance efforts.
- Data Archiving and Disaster Recovery: Robust data archiving strategies, coupled with comprehensive disaster recovery plans, are vital to ensure the long-term accessibility, integrity, and security of electronic records. This includes periodic verification of archived data.
- Regular Training and Competency Assessment: As systems and regulations evolve, ongoing personnel training is critical. Competency assessments can ensure that users understand their responsibilities regarding electronic records and signatures.
- Risk-Based Approach to Validation:As discussed with CSA, applying a risk-based approach to validation means focusing resources where they matter most and ensuring the level of validation effort is proportionate to the risk.
- SOPs and Documentation: Maintain a comprehensive suite of SOPs covering all aspects of electronic record and signature management, from user access and system administration to data backup and audit trail review. All compliance-related activities, including validation, training, and system changes, must be meticulously documented. This ensures audit readiness and provides a clear trail of compliance efforts. For a beginner’s guide to compliance, resources like Advarra’s blog can be helpful.
Essential Documentation for FDA Audit Readiness
When an FDA audit occurs, comprehensive and well-organized documentation is your first line of defense. Auditors will seek evidence of your compliance efforts across all aspects of 21 CFR Part 11. Key documents to have readily available include:
- Validation Plan and Report: These documents detail the scope of validation, testing protocols, test results, deviations, and a summary statement confirming the system’s fitness for its intended use.
- System Specifications: Detailed documentation of the electronic system’s design, functionality, and configuration.
- Standard Operating Procedures (SOPs): All relevant SOPs governing the use, maintenance, security, and data management of electronic systems and electronic signatures.
- User Training Records:Documentation of all training provided to personnel on the electronic system and Part 11 requirements.
- Audit Trail Review Records: Evidence of regular review of system audit trails.
- Change Control Records:Documentation of all changes made to the validated system.
- System Access Records: Records of user accounts, roles, and access permissions.
- Non-Repudiation Letters: If applicable, copies of letters sent to the FDA formally stating the organization’s intent to use electronic records and signatures and accepting their legal equivalence.
Having these documents organized and easily accessible is paramount for demonstrating compliance and avoiding FDA audit issues.
Navigating International Standards and EU Annex 11
While 21 CFR Part 11 governs explicitly FDA-regulated activities in the United States, clinical research is a global endeavor. Organizations operating internationally must also consider other regulations, notably EudraLex Volume 4 Annex 11, which applies to computerized systems in the European Union.
Annex 11 shares many similarities with 21 CFR Part 11, focusing on data integrity, audit trails, security, and validation. While there are differences in emphasis and specific requirements, validation activities performed under one regulation often meet the requirements of the other, especially when a risk-based approach is applied. Global harmonization efforts, such as those promoted by ICH GCP (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use Good Clinical Practice), also aim to standardize clinical trial conduct and data management practices worldwide. Understanding these interconnected regulations is crucial for global clinical research, as compliance with one often aids in compliance with others. For further reference on EU guidelines, consult EudraLex Volume 4 Annex 11.
Frequently Asked Questions about 21 CFR Part 11
We often encounter common questions from clinical research professionals grappling with Part 11. Here are answers to some of the most pressing:
What is the difference between a closed and an open system?
As discussed earlier, a closed system is one where the organization responsible for the content of electronic records also controls access to those records. This typically refers to internal, proprietary systems. An open system, conversely, is one where the content-responsible organization, such as cloud-based services or systems accessible over public networks, does not control access. Open systems generally require additional controls, such as encryption, to ensure data integrity during transmission.
Do all technology platforms used in research require full validation?
No, not all platforms require “full” (traditional, extensive) validation. The FDA promotes a risk-based approach. Validation efforts should be proportionate to the risk the system poses to patient safety, data integrity, and product quality. Systems that create, modify, maintain, archive, retrieve, or transmit records required by predicate rules, or those that use electronic signatures, will require some level of validation. However, the extent of that validation can vary significantly based on a thorough risk assessment. For example, a simple word processor used solely for internal SOPs might not require extensive validation, whereas an EDC system used for primary endpoint data would.
What are the consequences of using non-compliant electronic systems?
Non-compliance with 21 CFR Part 11 can lead to significant issues, including FDA audit findings, warning letters, and potentially severe regulatory penalties. This can result in delays in drug approvals, product recalls, and damage to an organization’s reputation. Crucially, non-compliant electronic records and signatures may not be accepted by the FDA, invalidating critical clinical trial data. This can lead to costly re-work, repeated studies, or even the rejection of regulatory submissions. Avoiding these issues requires proactive compliance planning, robust system validation, and continuous monitoring.
The Future of Regulatory Intelligence
The landscape of clinical research is dynamic, with technological advancements continually reshaping how we conduct studies. As AI, machine learning, and advanced analytics become more integrated into every facet of research, the need for intelligent regulatory compliance solutions will only grow.
The future of regulatory intelligence lies in leveraging these technologies to move beyond reactive compliance to a proactive, predictive model. AI can enable real-time monitoring of systems for compliance deviations, automate the generation of validation documentation, and even predict potential regulatory risks based on evolving guidance and historical data. This shift towards intelligent automation promises to make compliance more efficient, robust, and scalable, ultimately accelerating the delivery of safe and effective therapies to patients. By embracing AI-powered life sciences compliance, organizations can navigate the complexities of 21 CFR Part 11 and other regulations with greater agility and confidence, ensuring digital integrity in a rapidly evolving scientific world.





